GUI
Starting the GUI
The quickest way to start is to open the GUI from a command line terminal. You might need to open an anaconda prompt if you did not add anaconda to the path:
python -m cellpose
The first time cellpose runs it downloads the latest available trained model weights from the website.
You can drag and drop images (.tif, .png, .jpg, .gif) into the GUI and run Cellpose, and/or manually segment them. When the GUI is processing, you will see the progress bar fill up and during this time you cannot click on anything in the GUI. For more information about what the GUI is doing you can look at the terminal/prompt you opened the GUI with. For example data, see cellpose website. For best accuracy and runtime performance, resize images so cells are less than 100 pixels across.
For multi-channel, multi-Z tiff’s, the expected format is Z x channels x Ly x Lx.
For multi-Z 3D data, please use the 3D version of the GUI:
python -m cellpose --Zstack
Note
The output file with the masks is in the same folder as the loaded
image with _seg.npy appended. The GUI automatically saves after you
draw an ROI but NOT after running a model for
segmentation and NOT after 3D mask drawing (too slow). Save in the file
menu or with Ctrl+S.
Note
Since the output file is in the same folder as the loaded
image with _seg.npy appended, make sure you have WRITE access
in the folder, otherwise the file will not save.

Using the GUI
The GUI serves three main functions:
Running the segmentation algorithm.
Manually labelling data.
Fine-tuning a pretrained cellpose model on your own data.
Main GUI mouse controls (works in all views):
Pan = left-click + drag
Zoom = scroll wheel (or +/= and - buttons)
Select mask = left-click on mask
Delete mask = Ctrl (or Command on Mac) + left-click
Merge masks = Alt + left-click (will merge last two)
Start draw mask = right-click
End draw mask = press esc, right-click, or return to circle at beginning
Drawing masks
Masks are started with right-click, then hover your mouse (do not hold it down), and return it to the red circle to complete the mask. The mask should now be completed. At any point, you can escape drawing a mask with the Esc key.
Overlaps in masks are NOT allowed. If you draw a mask on top of another mask, it is cropped so that it doesn’t overlap with the old mask. Masks in 2D should be single strokes (if single_stroke is checked).
If you want to draw masks in 3D, then you can turn single_stroke option off and draw a stroke on each plane with the cell and then press ENTER. You can also draw multiple strokes on the same plane for complex cell shapes, but do not do this in 2D if you plan to train a cellpose model (the cell flows will not work correctly).
Note
3D labelling will fill in unlabelled z-planes so that you do not have to densely label, for example you can skip some planes, and the cell will be interpolated between planes.
After each mask is drawn in 2D, it is saved to the _seg.npy. If this is slow (for
large images), this “autosave” option can be turned off in the “File” menu
(“Disable autosave _seg.npy file”). In 3D,
the mask is never auto-saved, instead save masks by clicking CTRL+S, or “Save” in the
“File” menu.
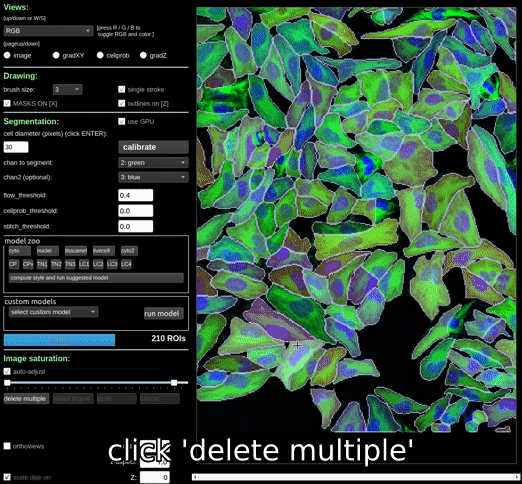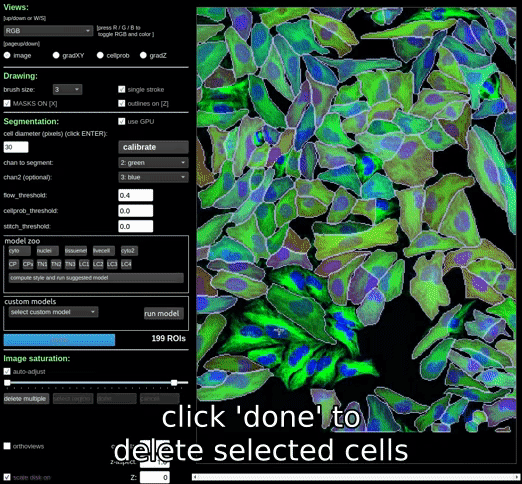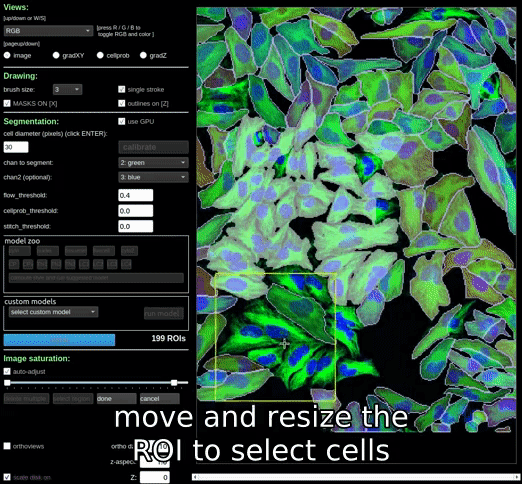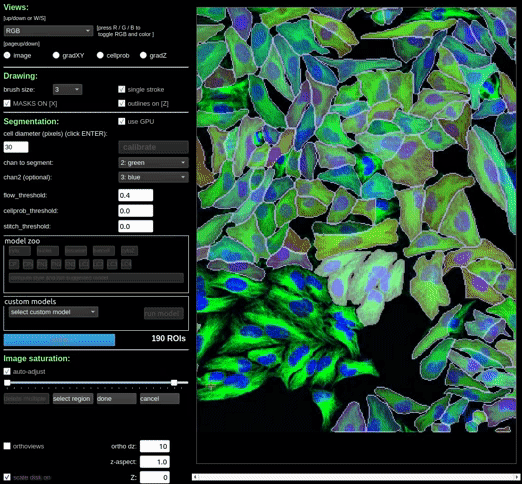
Bulk Mask Deletion
Clicking the ‘delete multiple’ button will allow you to select and delete multiple masks at once. Masks can be deselected by clicking on them again. Once you have selected all the masks you want to delete, click the ‘done’ button to delete them.
Alternatively, you can create a rectangular region to delete a regions of masks by clicking the ‘delete multiple’ button, and then moving and/or resizing the region to select the masks you want to delete. Once you have selected the masks you want to delete, click the ‘done’ button to delete them.
At any point in the process, you can click the ‘cancel’ button to cancel the bulk deletion.
Training your own cellpose model
Check out this video to learn the process.
Drag and drop an image from a folder of images with a similar style (like similar cell types).
Run the built-in models on one of the images using the “model zoo” and find the one that works best for your data. Make sure that if you have a nuclear channel you have selected it for CHAN2.
Fix the labelling by drawing new ROIs (right-click) and deleting incorrect ones (CTRL+click). The GUI autosaves any manual changes (but does not autosave after running the model, for that click CTRL+S). The segmentation is saved in a
_seg.npyfile.Go to the “Models” menu in the File bar at the top and click “Train new model…” or use shortcut CTRL+T.
Choose the pretrained model to start the training from (the model you used in #2), and type in the model name that you want to use. The other parameters should work well in general for most data types. Then click OK.
The model will train (much faster if you have a GPU) and then auto-run on the next image in the folder. Next you can repeat #3-#5 as many times as is necessary.
The trained model is available to use in the future in the GUI in the “custom model” section and is saved in your image folder.
If you have 3D data, please save random XY, YZ and XZ slices through your 3D data, ideally sufficiently spaced from each other so the information each slice has is distinct. Then put these slices into a folder and start the human-in-the-loop training. You can then use the new custom model on new 3D data.
Note
You can only start training with the built-in cpsam model. When you start training from a built-in model, then you are training the network on all the previously labelled images in the folder and weighting them equally in your training set.
If you restart from a previous retraining, you are biasing the network towards the earlier images it has already been trained on. Conversely, if you have created a custom model with different images, and you retrain that model, then you are downweighting the images that you have already trained on and excluded from your new training set. Therefore, we recommend having all images that you want to be trained for the same model in the same folder so they are all used.
See the Models doc for info on the new model zoo and suggestion mode.
Keyboard shortcuts
Keyboard shortcuts |
Description |
|---|---|
CTRL+H |
help |
=/+ // - |
zoom in // zoom out |
SPACE |
reset zoom |
CTRL+Z |
undo previously drawn mask/stroke |
CTRL+0 |
clear all masks |
CTRL+L |
load image (can alternatively drag and drop image) |
CTRL+S |
SAVE MASKS IN IMAGE to |
CTRL+T |
start model training using |
CTRL+P |
load |
CTRL+M |
load masks file (must be same size as image with 0 for NO mask, and 1,2,3… for masks) |
CTRL+N |
save masks as PNG |
CTRL+R |
save ROIs to native ImageJ ROI format |
CTRL+F |
save flows to image file |
A/D or LEFT/RIGHT |
cycle through images in current directory |
W/S or UP/DOWN |
change color (RGB/gray/red/green/blue) |
R / G / B |
press to toggle RGB and Red or Green or Blue |
PAGE-UP / PAGE-DOWN |
change to flows and cell prob views (if segmentation computed) |
X |
turn masks ON or OFF |
Z |
toggle outlines ON or OFF |
, / . |
increase / decrease brush size for drawing |